Computational PROTAC modeling is advancing quickly, but the field cannot compare methods reliably without shared
benchmarks, common molecular representations, consistent metrics, and transparent reporting.
Benchmarking is not about declaring one universal winner. It is about making methods comparable, reproducible, and
useful across targets, E3 ligases, linker chemotypes, and assay contexts. For PROTACs, that means evaluating both
geometry and downstream biological relevance without pretending one score can summarize everything.
Task-specific evaluationRepresentation mattersDomain shift mattersReproducibility first
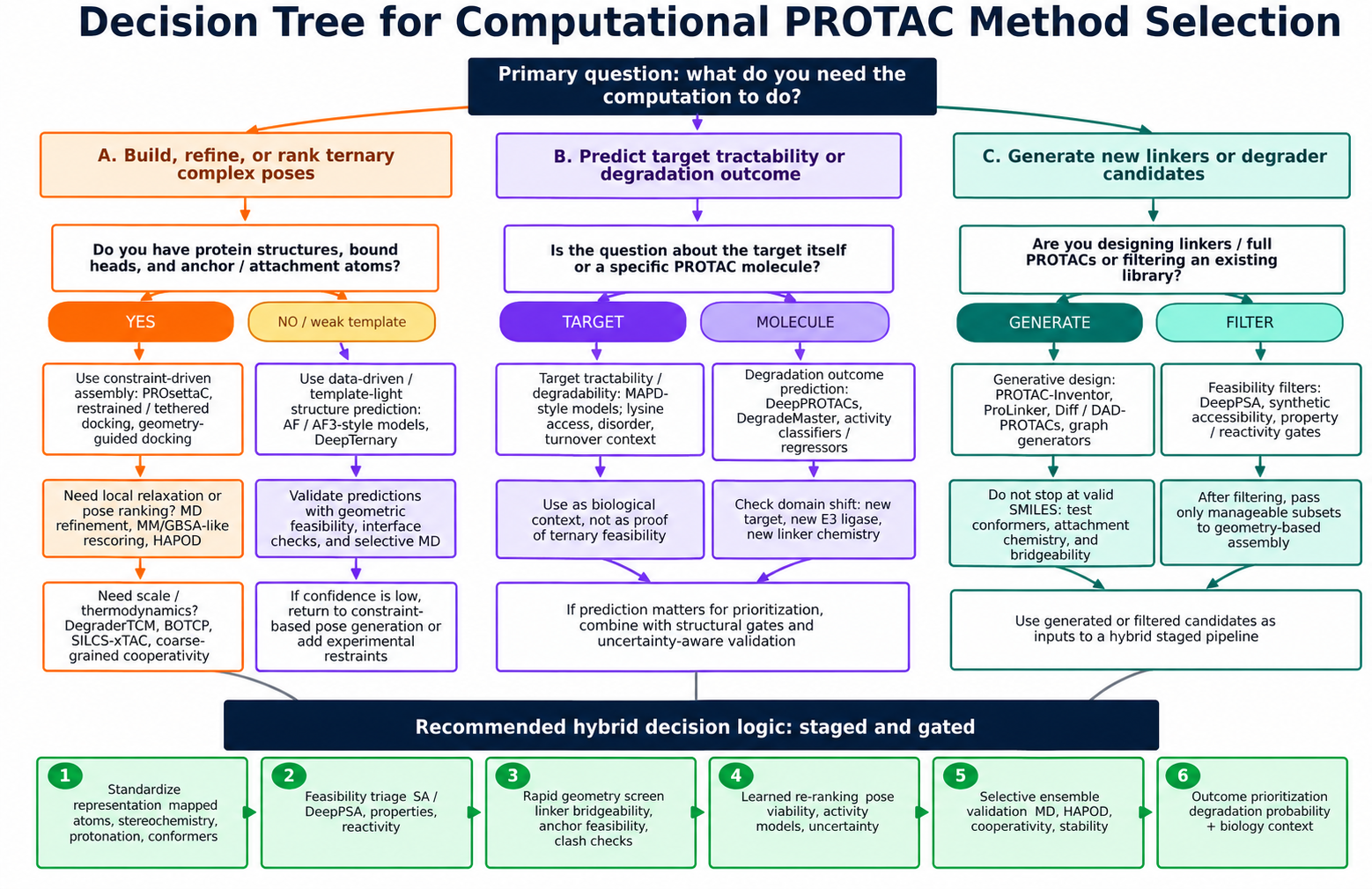
Figure 4. Decision framework for computational PROTAC workflows, showing how the modeling
objective determines whether users need structure prediction, outcome prediction, generative design, or staged
hybrid prioritization. Figure from the Schurer Lab in silico PROTAC modeling perspective draft, used here as
project-owned educational content.
Quick answer: what should a PROTAC benchmark measure?
A useful PROTAC benchmark should separate at least four tasks:
Ternary structure prediction: did the method recover the target-PROTAC-E3 geometry?
Pose ranking and enrichment: did the method rank productive poses above decoys?
Degradation outcome prediction: did the method predict degrader or non-degrader labels, DC50, Dmax, or selectivity?
Generative design evaluation: did generated candidates remain chemically valid, synthetically plausible, geometrically feasible, and experimentally testable?
Takeaway: no single score captures PROTAC success. Benchmarking should be task-specific,
multi-metric, and transparent about domain of applicability.
Why PROTAC benchmarking is hard
The key modeled object is a PROTAC-induced ternary complex, not a simple binary ligand-protein pose.
Degradation depends on more than static geometry, and different papers define “success” differently depending on
whether they are predicting structure, ranking poses, forecasting degradation, or generating chemistry.
Structural datasets are still small relative to PROTAC chemical and E3-ligase diversity.
Activity datasets are heterogeneous across assays, cell lines, targets, time points, and readouts.
High-quality negatives such as binders that do not degrade are often missing.
PROTAC molecules are large, flexible, and highly sensitive to molecular representation choices.
ML methods can look strong under random splits and then fail under harder target, E3, linker, or assay-context splits.
Community direction: the first goal is not to create one universal leaderboard too early. The
first goal is to define comparable tasks, inputs, outputs, and reporting rules.
Benchmarking tasks that should be kept separate
Ternary structure prediction
Question: can the method recover experimentally observed ternary complex geometry?
Metrics: DockQ, interface RMSD, ligand RMSD where applicable, fraction of native contacts, interface precision or recall, and clash or linker-feasibility checks.
Cautions: global alignment can hide degrader-relevant interface errors, accessory proteins can inflate scores, and static crystal structures are snapshots rather than full ensembles.
Pose ranking and enrichment
Question: can the method rank productive or near-native poses above decoys?
Metrics: top-1, top-5, or top-k success, enrichment factors, score calibration, rank correlation against reference quality, and decoy rejection rate.
Cautions: a method may generate a good pose but fail to rank it well, and raw docking or Rosetta energy should not be treated as uncalibrated truth.
Degradation outcome prediction
Question: can the method predict whether a candidate PROTAC degrades the target in a defined biological context?
Metrics: AUROC, AUPRC, accuracy, balanced accuracy, calibration curves, regression to DC50, Dmax, or percent degradation, and assay-stratified or target-E3 split performance.
Cautions: labels are noisy, assay context matters, and a plausible ternary structure does not guarantee cellular degradation.
Generative design evaluation
Question: do generated linkers or degraders survive downstream feasibility and validation filters?
Metrics: validity, uniqueness, novelty, synthetic accessibility, property distribution matching, conformer pass rate, bridgeability pass rate, modeling pass rate, and experimental follow-up rate when available.
Cautions: a molecule can be valid in 2D and still impossible or poor in 3D, so novelty is not enough without feasibility.
The case for a universal PROTAC benchmarking standard
A practical PROTAC benchmarking standard is still a proposed community direction rather than an established
standard. It should help groups compare results without forcing every method into the same mold.
What it should define
Common input schemas, common output formats, task-specific metrics, required metadata, recommended negative controls, domain-shift splits, and reproducibility assets.
What it should not claim
A universal standard does not mean one universal score. It means a shared language for what was modeled, how it was prepared, how it was evaluated, and where it should or should not be trusted.
Define the task.
Define the system.
Define the molecule.
Define the protein constructs.
Define the modeling protocol.
Define the metric.
Define the negative controls.
Define the domain split.
Release reproducibility assets.
State the domain of applicability.
Minimal benchmark dataset requirements
Structure-prediction benchmarks
Include PDB or structure source, POI and E3 chains, accessory proteins, PROTAC structure, ligand poses, anchor atoms, linker identity, construct boundaries, missing residues, biological assembly choice, and the scored interface definition.
Degradation-prediction benchmarks
Include target, E3, warhead, linker, recruiter structures, cell line, assay type, treatment time, concentration series, DC50 and Dmax when available, label definition, selectivity context, and inactive analogues when available.
Generative-design benchmarks
Include split definition, seed molecules or conditioning inputs, allowed chemistry constraints, validity and novelty metrics, downstream structural-feasibility metrics, and experimental confirmation status when available.
Negative controls are not optional
Benchmarks need binders that fail to degrade, linker-incompatible designs, nonproductive ternary poses, and decoy
poses that are chemically plausible but geometrically wrong. Without negatives, learned models can latch onto
shortcuts and pose-ranking methods cannot be evaluated fairly.
Same warhead and recruiter with an inactive linker
Linker too short or too long
Attachment atom that disrupts binding
Binary binders with no observed degradation
Ternary pose with severe linker strain
Target or E3 combination outside the training distribution
Molecular representation standards
PROTACs are representation-sensitive because they combine long flexible linkers, explicit attachment points,
stereochemistry, tautomer or protonation ambiguity, conformer-generation failures, and multiple possible head-group
definitions. Without consistent molecular and workflow representations, it becomes difficult to compare docking,
learned scoring, and generative outputs in a reproducible way.
Canonical 2D encoding with explicit attachment-point labels.
Atom-mapped SMILES or equivalent connection-point notation.
Stereochemistry specified.
Protonation and tautomer state specified or documented.
Warhead, linker, and E3 recruiter boundaries defined.
Anchor atoms defined.
3D conformer protocol documented.
Failed conformer-generation cases reported.
Protein target and E3 metadata linked to the molecule.
Assay context included when degradation labels are used.
Builder role: PROTAC Builder can help users standardize component boundaries, attachment atoms,
linker choices, and exportable representations before downstream modeling or benchmarking.
Recommended reporting checklist
To improve reproducibility, cross-method comparison, and prospective usability, computational PROTAC studies should
report the following whenever applicable.
1. System definition
Protein structures, constructs, chain definitions, modeled domains, and E3 complex composition.
2. PROTAC representation
Warhead and E3-ligand binding modes, linker attachment atoms, anchor definitions, stereochemistry, protonation, and tautomer states.
Definitions of pose success, ranking success, degradation-prediction success, generative-model success, top-k metrics, and enrichment metrics.
6. Negative and control examples
Binders that fail to degrade, linker-incompatible designs, nonproductive ternary poses, and other control cases.
7. Reproducibility assets
Random seeds, model checkpoints, scripts, containers, benchmark inputs, and output files needed to reproduce the workflow.
8. Domain of applicability
Limitations for new targets, E3 ligases, linker chemotypes, molecular-glue-like systems, or biological contexts outside the validation set.
Domain shift in PROTAC modeling
Random splits can overestimate performance. The field needs harder generalization tests that expose where methods
collapse rather than only where they succeed.
Leave-one-target-family-out
Tests whether the model can move beyond familiar protein families.
Leave-one-E3-ligase-out
Tests whether performance generalizes beyond dominant CRBN or VHL-heavy datasets.
Leave-one-warhead-chemotype-out
Tests learned dependence on familiar POI-ligand chemistry.
Leave-one-linker-class-out
Tests generalization beyond common PEG or alkyl linker motifs.
Temporal and scaffold splits
Reduce leakage from near-duplicate analogues or chronologically later structures.
Assay-context splits
Expose dependence on one cell line, one readout format, or one treatment window.
Practical takeaway: a benchmark that only tests familiar CRBN or VHL systems does not prove
generalization to the broader degradome.
Scoring: why one number is not enough
Geometry and stability do not uniquely determine degradation. DockQ or RMSD may help for structure recovery but do
not measure cellular activity. Docking or Rosetta energy may not correlate with interface correctness. ML confidence
may not be calibrated under domain shift. Generative validity does not mean biological usefulness.
Gate 1: chemical validity and representation sanity
Gate 2: geometric feasibility and linker bridgeability
Gate 3: interface plausibility and clash or contact checks
Gate 4: ensemble stability or dynamic persistence
Gate 5: ubiquitination-plausibility proxies
Gate 6: learned degradation prediction with uncertainty
Gate 7: experimental degradation and selectivity validation
Proposed universal benchmark output schema
This is a conceptual example, not a backend implementation requirement. The goal is to show the kind of structured
metadata that makes benchmark outputs comparable across groups.
Curate small but high-quality task-specific benchmark sets.
Define common molecular representation rules.
Create structure-prediction benchmarks with clear interface scoring.
Create pose-ranking benchmarks with realistic decoys.
Create degradation-prediction benchmarks with well-defined labels and negatives.
Create generative-design benchmarks that measure downstream pass rates.
Require domain-shift splits.
Publish scripts, containers, and input or output examples.
Report failure modes as first-class results.
Connect benchmark inputs to practical assembly tools like PROTAC Builder.
How PROTAC Builder supports benchmark-ready workflows
PROTAC Builder is not a benchmark standard and not a validated benchmarking engine. It can, however, serve as a
preparation and standardization layer for benchmark-ready computational workflows by helping users normalize
component boundaries, attachment atoms, and assembly outputs before downstream modeling.
What it can standardize
Component selection, warhead and recruiter boundaries, attachment atoms, linker choices, assembled candidate structures, export formats, and batch or API-ready handoffs.
What it can'tclaim
It is not presented as the official global benchmark standard or as a guarantee of predictive modeling performance or biological degradation.
This page is based in part on the Schurer Lab perspective draft From Ternary Modeling to Predictive PROTAC
Design: A Computational Perspective. It is intended as an educational benchmarking guide & will be maintained by the lab and team.