Building a PROTAC is not just joining two ligands with a spacer. The molecule must engage the protein of interest,
recruit an E3 ligase, support a productive ternary complex, position the target for ubiquitination, enter cells,
and avoid an unmanageable physicochemical burden.
This guide turns that challenge into a staged workflow for designing, assembling, prioritizing, and validating
candidate degraders without overstating what any single computational step can prove.
PROTAC design workflowAttachment-aware designTernary-complex focusedValidation still required
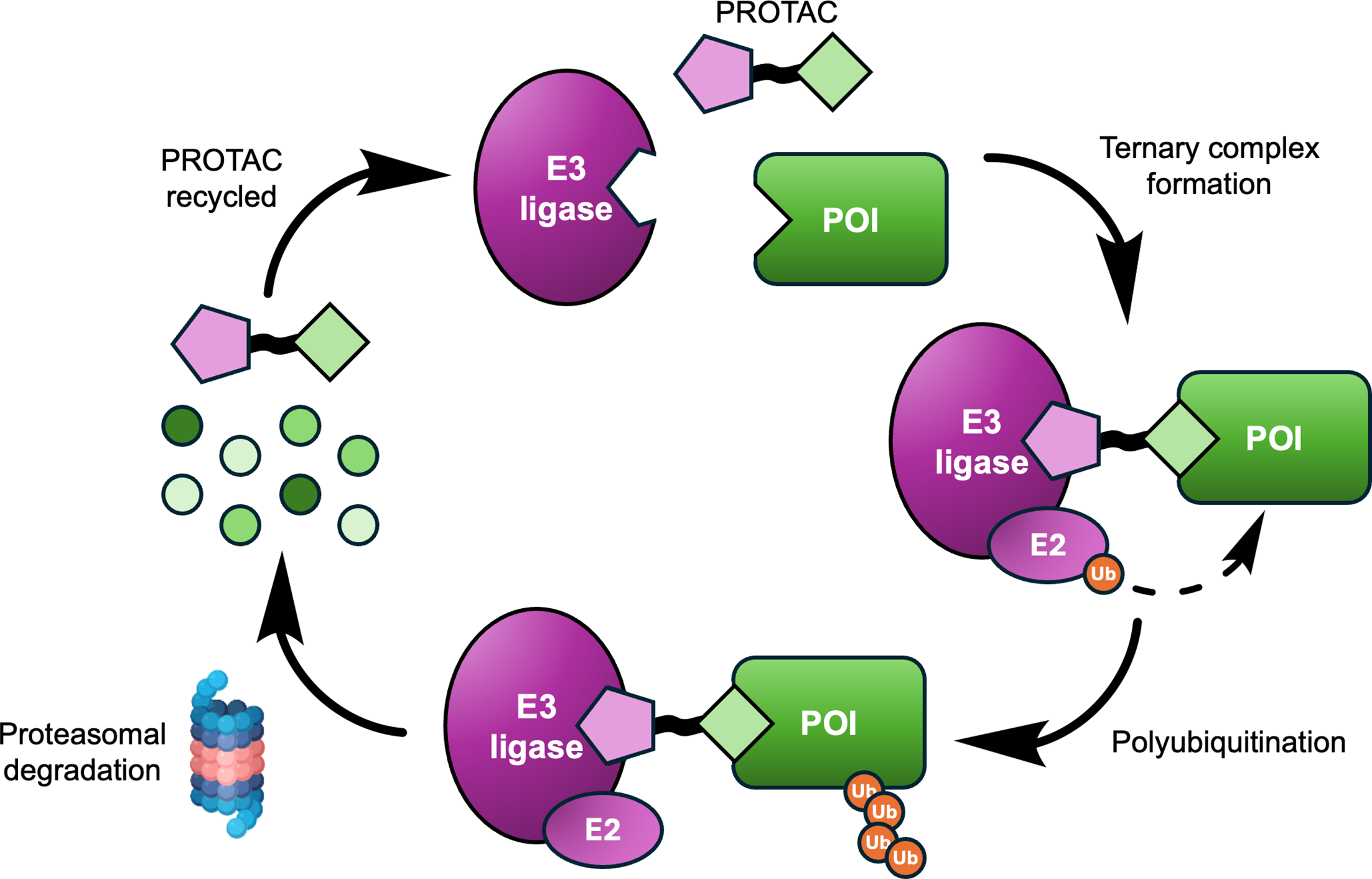
Figure 1. Mechanistic overview: a PROTAC brings a POI into proximity with an E3 ligase, enabling ubiquitin transfer, proteasomal degradation, and potential PROTAC recycling.
Source: Osman, Thompson, Jörg, and Scanlon, Biochemical Journal (2025),
doi:10.1042/BCJ20243018.
Quick answer: how do you build a PROTAC?
Pick a protein of interest and confirm that degradation is biologically meaningful.
Choose or discover a target-binding warhead.
Select an E3 ligase recruiter, often starting with VHL or CRBN when appropriate.
Identify solvent-exposed attachment vectors on both ligands.
Choose a linker panel that varies length, polarity, and rigidity.
Assemble candidate PROTACs.
Prioritize candidates with descriptor checks, structural review, and downstream modeling where useful.
Test degradation in cells and interpret DC50, Dmax, selectivity, and hook effect.
Step 1: understand the PROTAC mechanism
A PROTAC usually contains three modular parts: a ligand for the protein of interest, a linker, and an E3 ligase recruiter.
The degrader works only if those parts support a productive ternary complex rather than two disconnected binary interactions.
The POI ligand binds the target protein.
The E3 recruiter binds an endogenous E3 ligase.
The ternary complex enables E2 or E3-mediated ubiquitin transfer to the POI.
Polyubiquitinated POI is sent to the proteasome for degradation.
The PROTAC may then dissociate and engage another target molecule.
Why this matters: strong occupancy alone is not the end goal. The real objective is productive target degradation in a relevant cellular setting.
Figure 1. The workflow begins with ternary complex formation and ends with degradation rather than simple target occupancy.
Source: Osman et al., Biochemical Journal (2025),
doi:10.1042/BCJ20243018.
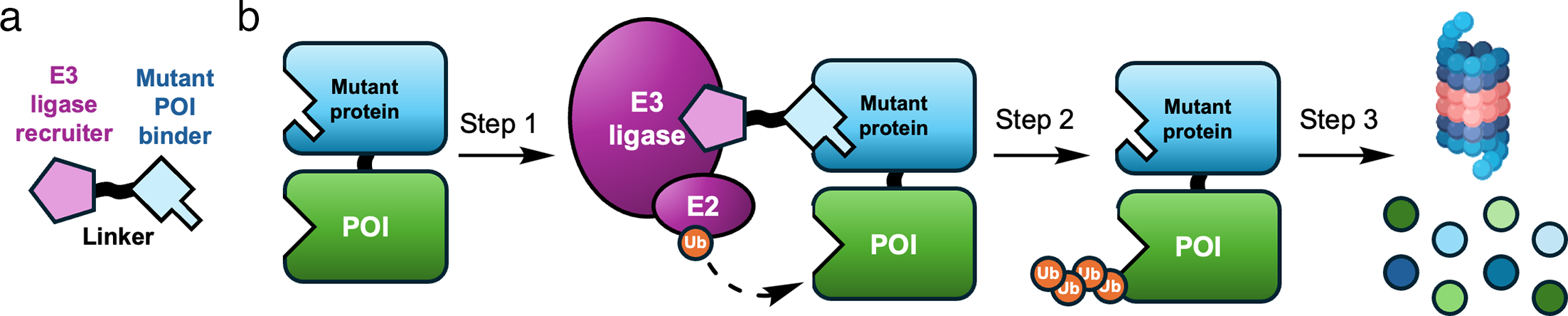
Step 2: choose the right protein of interest
Figure 2. Disease-context or mutant-selective degradation can be valuable when the biology calls for precision beyond broad target occupancy.
Source: Osman et al., Biochemical Journal (2025),
doi:10.1042/BCJ20243018.
A good PROTAC project starts with target selection, not linker shopping. The POI should have a strong biological rationale and a reason why degradation may be more informative or more therapeutic than inhibition alone.
Degradation can remove scaffolding or non-enzymatic functions that inhibitors do not fully address.
Mutant-selective or disease-selective contexts may matter for safety, selectivity, or therapeutic rationale.
The best binder does not always need to hit an active site if it can support productive ternary geometry.
Cellular localization, expression level, turnover rate, lysine accessibility, and assay availability all influence feasibility.
Sanity check: before designing chemistry, ask whether losing the entire protein is actually the desired biological intervention.
Step 3: choose a protein-of-interest ligand or warhead
Warhead selection usually starts from known ligands, inhibitors, fragments, covalent binders, literature probes, or structure-enabled hits that already engage the POI. The goal is not just affinity, but a ligand that can tolerate derivatization without losing the interactions that matter.
Is the binding affinity or engagement quality sufficient for a starting point?
Is the binding site compatible with a degradation mechanism rather than only inhibition?
Is there a solvent-exposed exit vector or derivatization handle?
Could derivatization damage key hydrogen bonds, hydrophobic contacts, or selectivity features?
Is the target intracellular and compatible with proteasome-dependent degradation workflows?
Are there selectivity liabilities you should understand before making a degrader series?
Upstream Discovery
Warhead Hunter
Use the sister platform when the target-binding ligand is still under exploration or needs more target-context analysis.
The E3 recruiter determines which ligase you bring into the ternary complex and therefore influences geometry, cooperativity, degradation profile, and tissue or cell-context suitability.
VHL and CRBN are common starting points, but not universal answers.
E3 choice should be considered alongside cell type, tissue expression, biology, and known off-target liabilities.
Recruiter geometry can strongly reshape which ternary complexes are accessible.
If recruiter-side uncertainty is high, inspect recruiter scaffold context and ligase-side attachment options before you spend too much effort on linker enumeration.
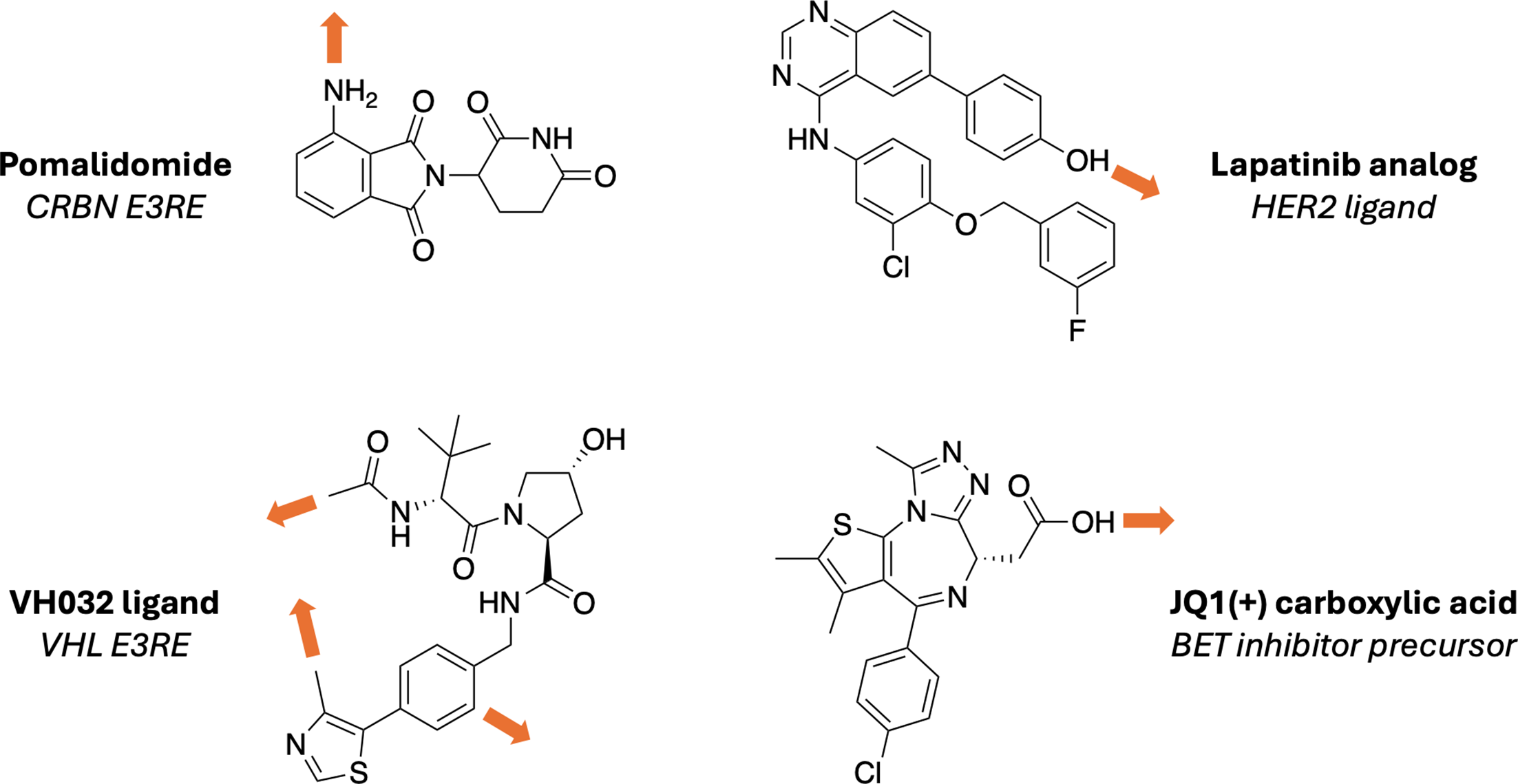
Figure 3. Representative recruiter and warhead building blocks, along with plausible attachment-vector positions, illustrate how component choice and derivatization strategy are linked.
Source: Osman et al., Biochemical Journal (2025),
doi:10.1042/BCJ20243018.
Recruiter Discovery
E3 Ligandalyzer
Inspect recruiter scaffolds, ligase context, and recruiter-side attachment ideas before assembly.
Both the POI ligand and the E3 recruiter need plausible attachment atoms or exit vectors. A bad exit-vector choice can preserve binary binding and still fail because the resulting ternary geometry is unproductive.
Prefer solvent-exposed positions supported by structures, SAR, docking hypotheses, or prior literature.
If uncertainty is high, test more than one attachment vector instead of assuming a single best exit point.
Document why each vector was selected so the design logic survives later rounds of iteration.
Practical rule: treat exit-vector selection as part of the degrader hypothesis, not as a small medicinal-chemistry detail to postpone.
Step 6: select linker length and exit vectors carefully
Do not assume one linker is best. Start with a small panel that varies length, flexibility, polarity, rigidity, and attachment chemistry so you can learn how the POI and recruiter tolerate different bridge geometries.
Flexible linkers
Useful early because they are easy to vary, but can add entropy and metabolic liability.
Rigid linkers
May preserve geometry and lower entropic cost, but can fail quickly when vectors are misaligned.
PEG-rich linkers
Can improve solubility but may increase polarity and limit permeability.
Alkyl and triazole motifs
Alkyl motifs are simple and tunable; triazole or click-enabled motifs are useful for modular assembly and geometric control.
Figure 3. The same component view is also an attachment-vector reminder: recruiter and POI ligands need derivatization sites that support a productive bridge.
Source: Osman et al., Biochemical Journal (2025),
doi:10.1042/BCJ20243018.
Component Guide
Linker design guide
Read the deeper linker-design hub before committing to a narrow linker strategy.
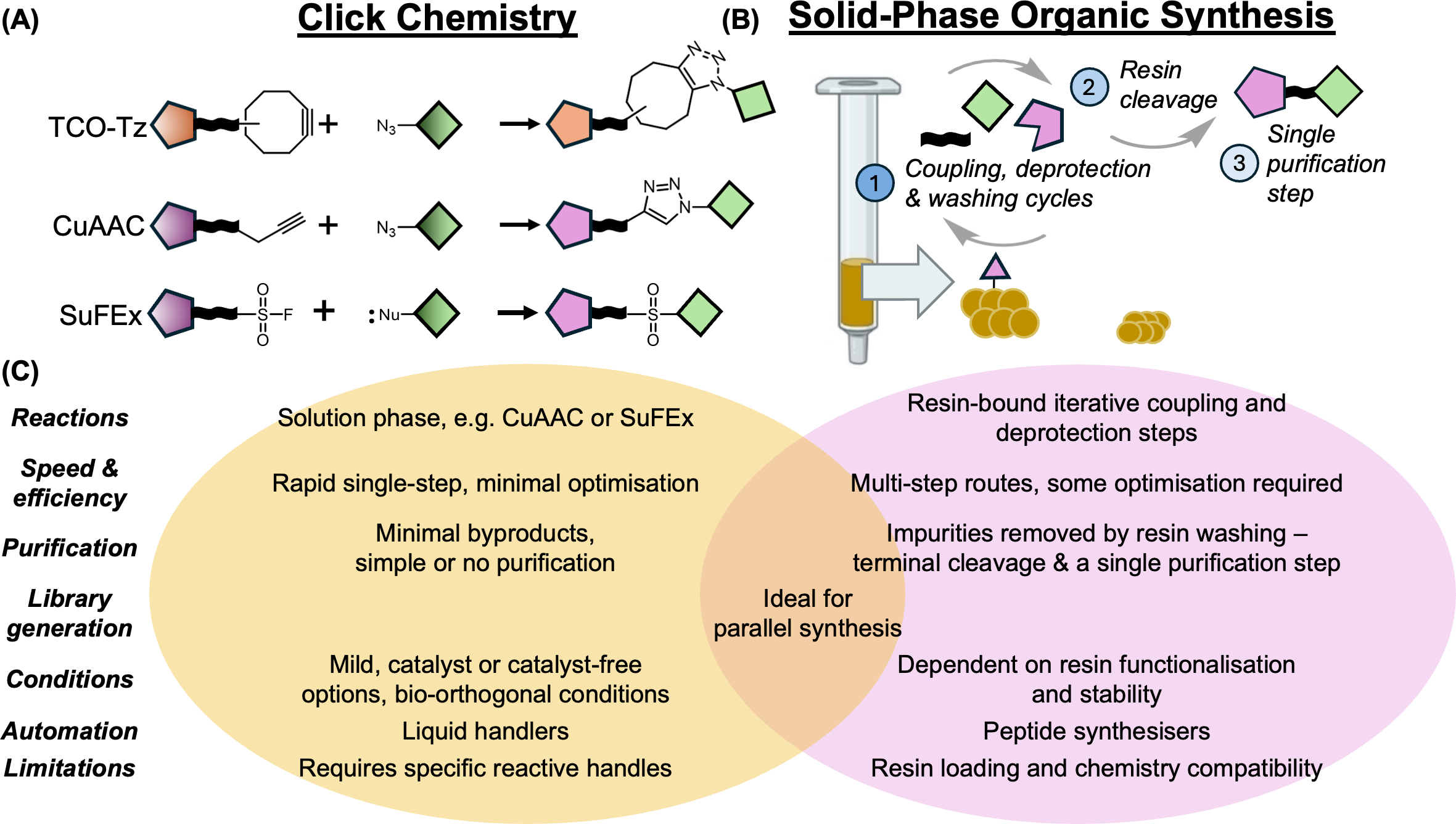
Figure 4. Modular assembly strategies such as click-enabled workflows, CuAAC, SuFEx, and solid-phase iteration can accelerate analogue generation when compatible handles are available.
Source: Osman et al., Biochemical Journal (2025),
doi:10.1042/BCJ20243018.
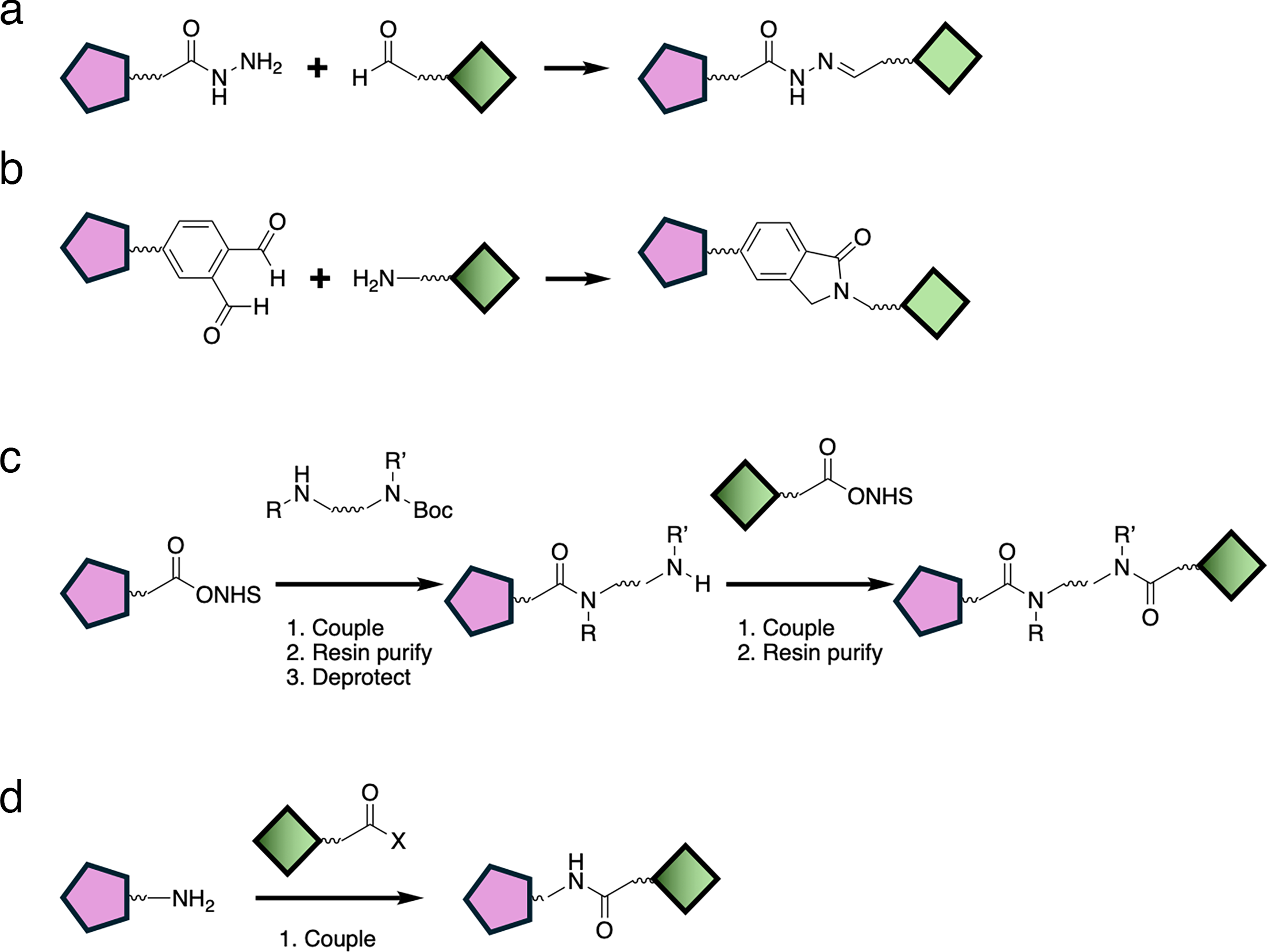
Figure 7. Representative coupling schemes reinforce the idea that chemistry strategy should match the available handles, stability constraints, purification needs, and desired iteration speed.
Source: Osman et al., Biochemical Journal (2025),
doi:10.1042/BCJ20243018.
From a workflow perspective, PROTAC synthesis is usually easier to scale when the design is modular. High-level choices like click chemistry, CuAAC, SuFEx, or solid-phase iterations can help accelerate analogue generation, but the right route depends on the available functional handles and downstream purification realities.
PROTAC Builder fits here by standardizing component selection, candidate assembly, linker choice, and exportable representations for later modeling or batch workflows. It does not replace chemistry development, but it does make the design space easier to organize and compare.
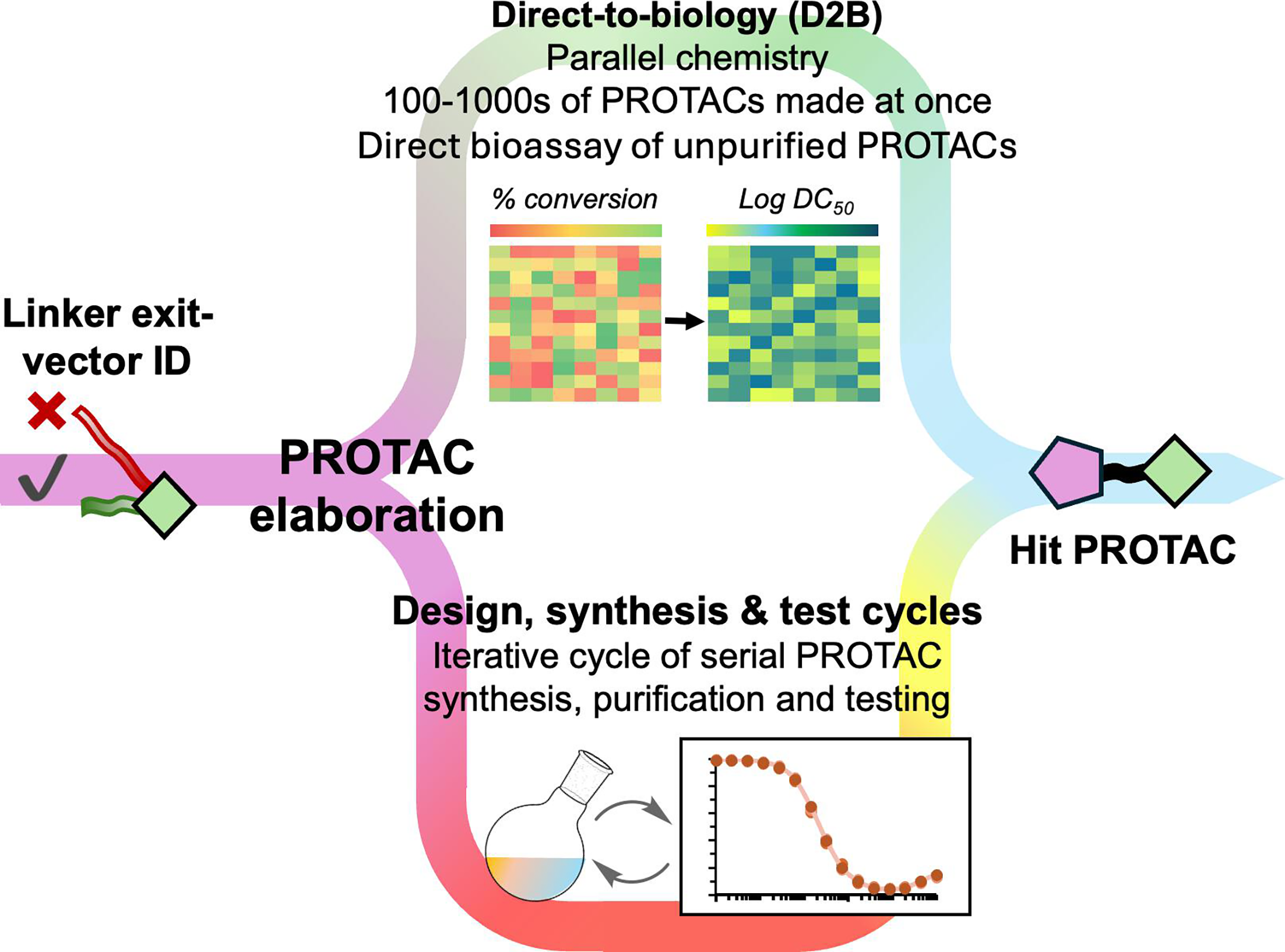
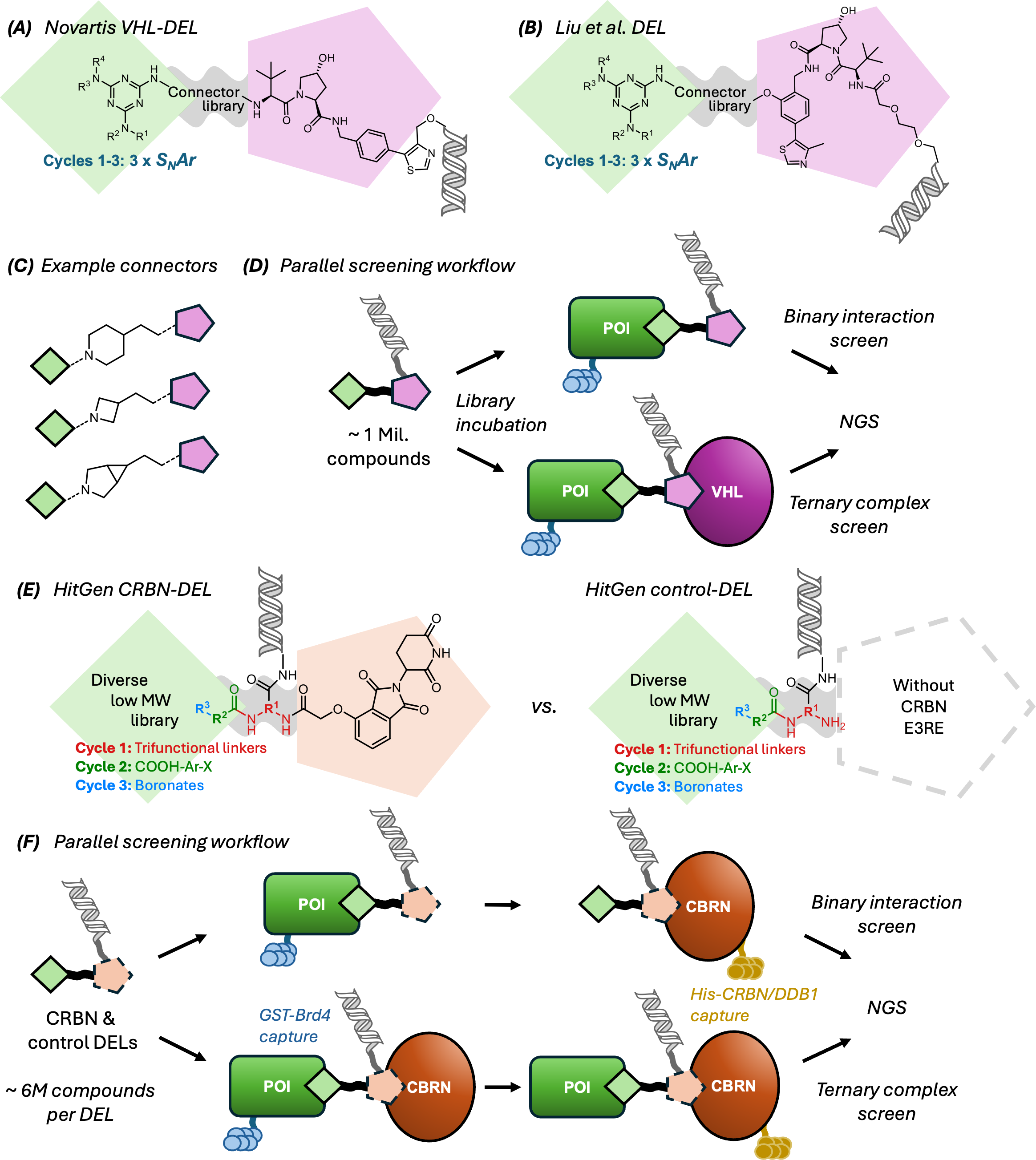
Step 8: scale exploration with libraries and batch workflows
Figure 5. DNA-encoded and parallel screening workflows can explore very large degrader spaces, and binary versus ternary screens answer different questions about what a candidate is doing.
Source: Osman et al., Biochemical Journal (2025),
doi:10.1042/BCJ20243018.
Acceleration often comes from scale. DNA-encoded libraries, connector libraries, and batch workflows can help probe many POI or recruiter combinations much faster than serial one-off chemistry.
Binary binding screens tell you whether one side of the molecule still engages.
Ternary complex screens ask a different, often more relevant, question.
Batch or API workflows help standardize larger candidate enumerations before synthesis or testing.
Scale Up
API Builder
Operationalize larger candidate sets once you know what component combinations are worth exploring.
Check structural validity and attachment-vector sanity first.
Review molecular weight, cLogP or logD, HBD/HBA count, TPSA, and rotatable bonds.
Flag permeability and solubility risk early, especially for large or highly polar scaffolds.
Think about synthetic feasibility, not just model quality.
Avoid over-filtering by classic small-molecule rules alone because PROTACs often sit beyond Rule-of-Five space.
Computational prioritization can help, especially when you have too many candidates to test immediately. Useful downstream methods include docking, restrained ternary modeling, molecular dynamics, and structure-guided refinement, but they should rank hypotheses rather than declare biological truth.
Caution: a visually convincing ternary pose is still only a model until degradation data, selectivity data, and exposure-aware interpretation support it.
Modeling
Downstream modeling tools
See how assembled candidates can move into restrained ternary modeling, docking, and scoring workflows.
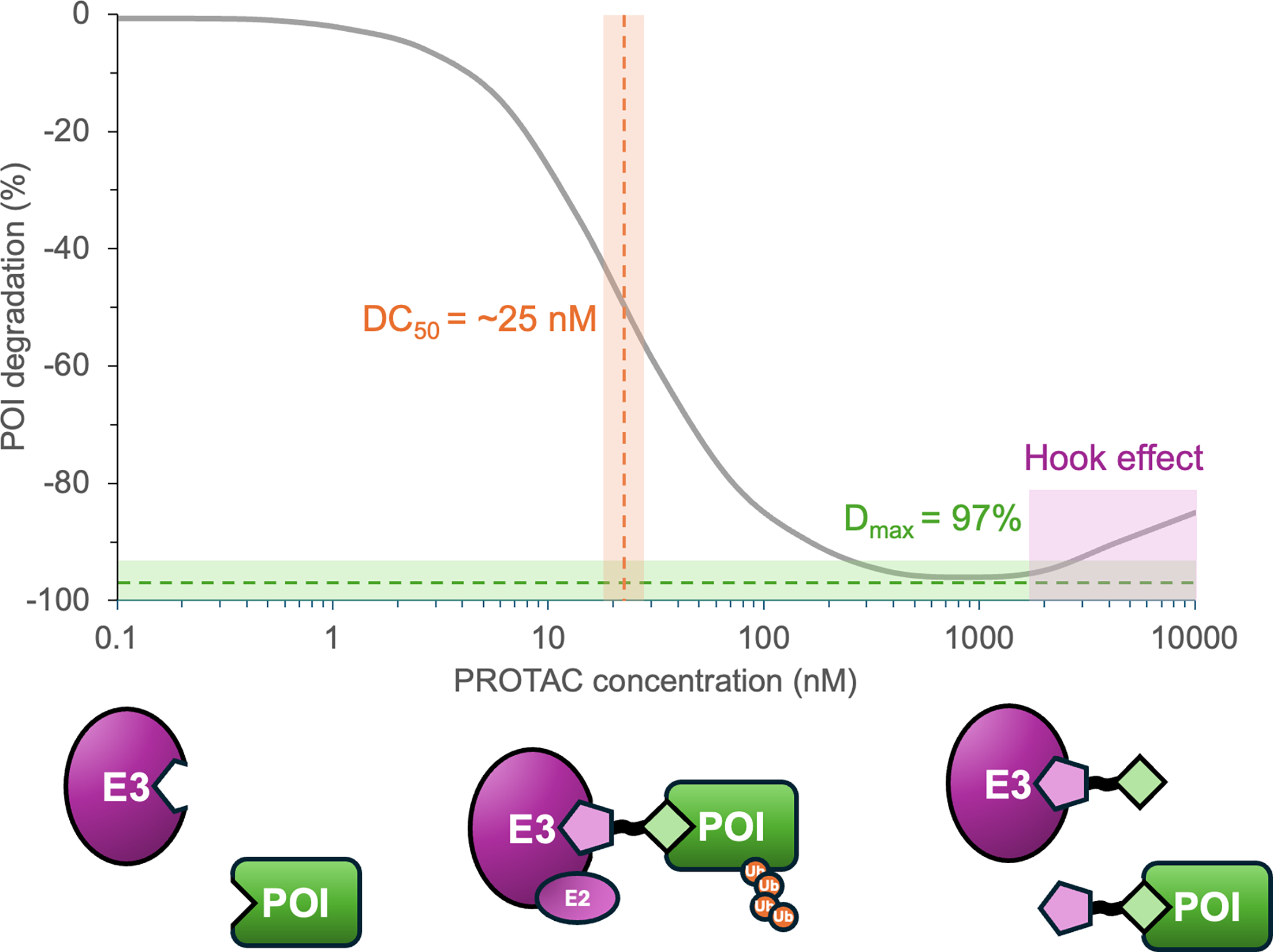
Step 10: measure degradation: DC50, Dmax, and hook effect
PROTACs are evaluated by degradation behavior, not only by occupancy. A useful degrader needs enough cellular engagement to produce degradation, enough maximal effect to matter biologically, and enough selectivity to support interpretation.
DC50: concentration associated with half-maximal degradation.
Dmax: maximum observed degradation.
Hook effect: reduced degradation at high PROTAC concentrations when excess binary complexes compete with productive ternary complex formation.
At a high level, degradation can be followed by Western blot, targeted proteomics, reporter systems, luminescence-style readouts, or broader cellular degradation assays depending on the project.
Interpret carefully: strong POI binding does not guarantee degradation, and modest binary binding can still work if ternary cooperativity is favorable.
Figure 8. Degradation-response curves should be read in terms of potency, maximal degradation, and high-dose behavior rather than a single concentration or a single headline number.
Source: Osman et al., Biochemical Journal (2025),
doi:10.1042/BCJ20243018.
Step 11: use direct-to-biology carefully to shorten design cycles
Figure 6. Direct-to-biology can speed trend-finding by testing many crude or minimally purified analogues in parallel when the chemistry and assay format are compatible.
Source: Osman et al., Biochemical Journal (2025),
doi:10.1042/BCJ20243018.
Direct-to-biology is best thought of as a discovery accelerator. It can reveal exit-vector trends, linker trends, and promising hit PROTACs faster by coupling parallel chemistry with rapid biological readouts.
It is useful for trend detection, not final proof.
Assays must be robust enough to tolerate mixtures and impurity-related noise.
Purified compound confirmation is still required before strong conclusions are drawn.
Practical design checklist
POI is biologically justified.
POI has a ligand or warhead worth elaborating.
E3 recruiter is selected with cell context in mind.
POI-side attachment vector is plausible.
Recruiter-side attachment vector is plausible.
Linker panel varies length and properties instead of relying on one guess.
Candidate structures are chemically valid.
Physicochemical burden is being watched, not ignored.
A degradation assay plan exists.
DC50, Dmax, selectivity, and hook effect will all be evaluated.
A downstream export or modeling path is clear.
Computational prioritization will be confirmed experimentally.
Common mistakes to avoid
Treating the linker as a passive spacer.
Optimizing binary affinity only.
Ignoring ternary complex geometry.
Choosing attachment atoms that disrupt binding.
Testing too few linker lengths.
Ignoring permeability and solubility risks.
Over-trusting docking without experiments.
Ignoring E3 expression and biology.
Misreading hook effect as simple inactivity.
Comparing compounds only by DC50 without Dmax and selectivity context.
Recommended tool path
Upstream Discovery
Warhead Hunter
Upstream POI ligand discovery and target-context exploration.
Primary review used to ground this page:
Osman J, Thompson PE, Jörg M, Scanlon MJ.
“Methods to accelerate PROTAC drug discovery.”
Biochemical Journal, 2025; 482(13): 921-937.
Figures on this page are local copies derived from the cited review and are shown with visible attribution so readers can find the original paper for full context and license terms.
This guide is educational and workflow-oriented. It is grounded in the cited review and established PROTAC design principles, but it should still be reviewed by a domain expert before being used as publication-quality scientific guidance.